1. Disease overview
Congenital lipoid adrenal hyperplasia (LCAH) is a disorder characterized by deficiency of all steroid hormones produced by the adrenal glands and gonads due to dysfunction of steroidogenic acute regulatory protein (StAR), with hyperplasia of steroidogenic cells and accumulation of lipid droplets in the cytoplasm. In 46,XY individuals, this condition is also classified as a disorder of sex development that can manifest with atypical external genitalia. In Japan, it is the second most common form of congenital adrenal hyperplasia after 21-hydroxylase deficiency, accounting for approximately 4% of cases.
2. Etiology
LCAH is an autosomal recessive disorder caused by biallelic loss-of-function pathogenic variants in the STAR gene encoding StAR. Because the p.Glu258* variant in the STAR gene has a founder effect in East Asia, this condition is common in Japanese, Korean, and Chinese populations, and very rare in Caucasians.
3. Pathophysiology
StAR is strongly expressed in all steroidogenic cells of the adrenal glands and gonads. StAR transfers cholesterol from the outer to the inner mitochondrial membrane in response to demand for steroid hormones. This supply of cholesterol to the inner mitochondrial membrane is the rate-limiting step common to all steroid hormone biosynthesis. The primary pathophysiology of LCAH is the inability to rapidly supply cholesterol to the inner mitochondrial membrane, preventing production of steroid hormones commensurate with demand. Under stimulation by trophic hormones, steroidogenic cells of the adrenal glands and gonads show hyperplasia and accumulate lipid droplets in the cytoplasm, primarily consisting of cholesterol esters.
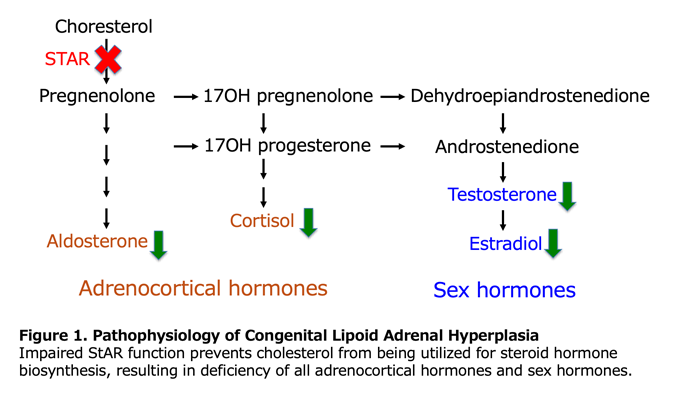

4. Clinical manifestations
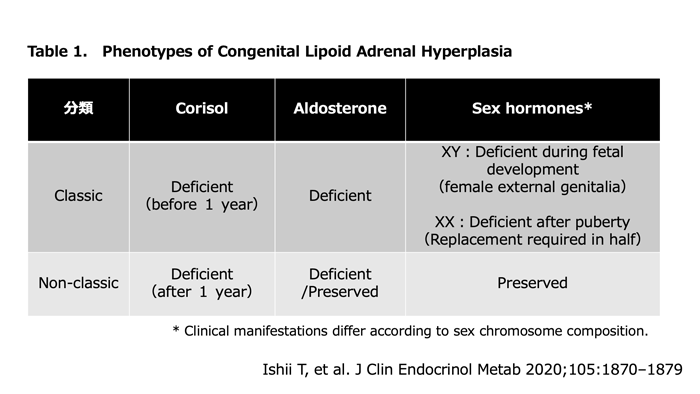
This condition is divided into two phenotypes: classic and non-classic. In Japan, 80% of cases are classic and 20% are non-classic.
1) Classic form:
The classic form is relatively severe, presenting with deficiency of all adrenocortical hormones and developing adrenal insufficiency in the neonatal or early infancy period. In 46,XY cases, the external genitalia are female type. In 90% of 46,XX cases, breast development and menarche occur spontaneously without delay. However, menstrual irregularity often develops subsequently, leading to premature ovarian failure.
2) Non-classic form:
The non-classic form is defined when adrenal insufficiency manifests after age 1 year, mineralocorticoid secretion is preserved, or 46,XY cases have completely male external genitalia. Mild hypergonadotropic hypogonadism is seen in some cases of both sexes.
5. Diagnosis and testing
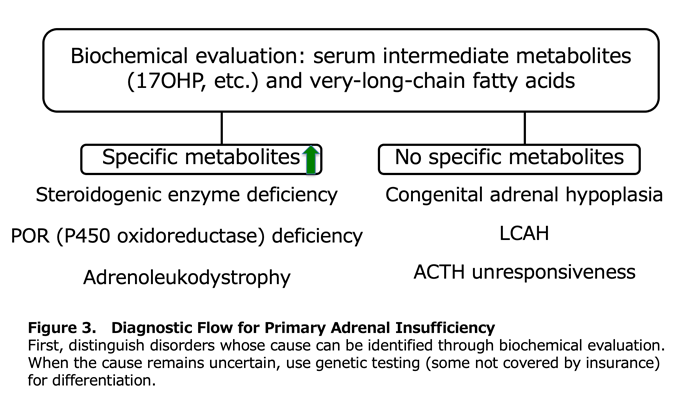
1) Diagnostic Flow for Primary Adrenocortical Insufficiency (Figure 3)
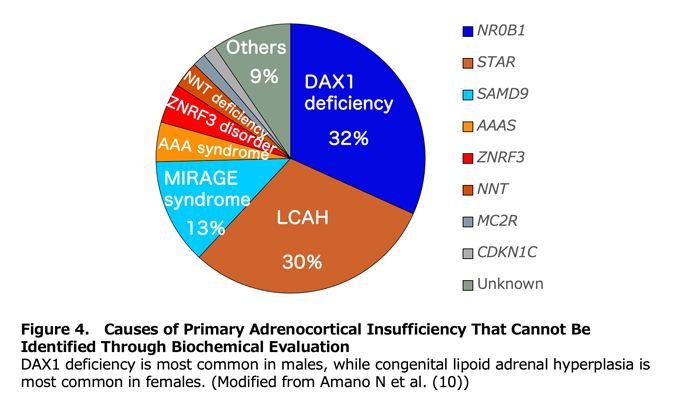
Primary adrenocortical insufficiency is diagnosed when clinical symptoms occur alongside elevated plasma ACTH and renin. The specific disorder is then identified through biochemical evaluation of intermediate metabolites such as 17-hydroxyprogesterone (17OHP) and very-long-chain fatty acids. Congenital lipoid adrenal hyperplasia (LCAH) lacks distinctive biochemical features; in the absence of characteristic gonadal phenotypes, it cannot be reliably distinguished from congenital adrenal hypoplasia or ACTH unresponsiveness. Among cases of primary adrenocortical insufficiency in which biochemical evaluation does not establish a cause, LCAH is the most common disorder in females (Figure 4).
2) Diagnosis of LCAH
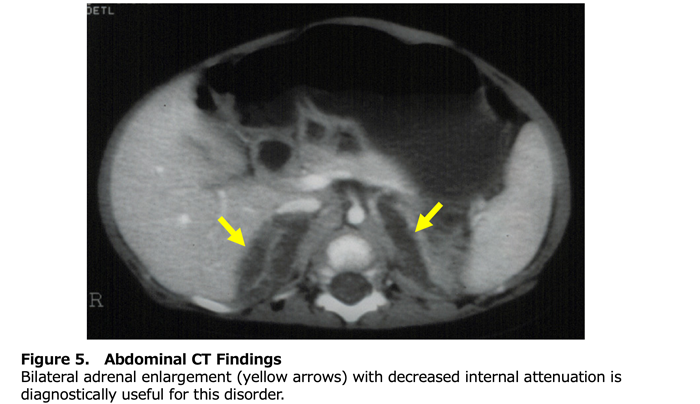
Table 2 presents the diagnostic criteria for this designated intractable disease. Classical LCAH should be suspected when all adrenal and gonadal steroid hormones are low, plasma ACTH, plasma renin, and serum gonadotropins are all elevated, and external genitalia are completely female in appearance. As with other forms of primary adrenocortical insufficiency, routine laboratory studies should assess for hyponatremia, hyperkalemia, and hypoglycemia. Abdominal computed tomography (CT) is the most informative imaging modality. Enlarged adrenal glands with decreased CT attenuation due to lipid accumulation (fatty attenuation) are seen in 86% of classical cases and are specific to LCAH (Figure 5) (7). However, the absence of adrenal enlargement does not exclude the diagnosis. In 46,XY individuals, ultrasound can identify testes along the path from the scrotum or labioscrotal folds through the inguinal canal. Genetic analysis of the STAR gene is highly valuable for diagnosis. It is particularly important for distinguishing LCAH from cholesterol side-chain cleavage enzyme deficiency when adrenal enlargement is absent, and for differentiating classical or non-classical LCAH from ACTH unresponsiveness in 46,XX individuals with preserved ovarian function or in 46,XY non-classical cases with preserved testicular function.

6. Treatment
1) Adrenocortical hormones:
Replacement therapy is administered according to the protocol for adrenal hypoplasia. Hydrocortisone replacement is necessary in almost all cases. Fludrocortisone replacement is necessary in all classic cases and 63% of non-classic cases. Target doses are hydrocortisone 10-25 mg/m2/day in neonatal-infancy period, 10-15 mg/m2/day from early childhood onward, and fludrocortisone 0.025-0.2 mg/day. During physical stress, it is necessary to increase hydrocortisone to 3-4 times the usual dose or 50-100 mg/m2/day.
2) External genitalia surgery and sex hormones:
In classic and non-classic 46,XY cases, male or female external genitalia surgery is often performed to match the assigned sex. In females with bilateral orchiectomy, estrogen replacement is performed at pubertal age. In classic 46,XX cases, estrogen replacement becomes necessary when secondary sexual characteristics do not progress or when premature ovarian insufficiency occurs.
7. References
- Stocco DM, Clark BJ. Endocr Rev 1996; 17: 221-244
- Ministry of Health, Labour and Welfare Research, 2010 National Survey
(http://www.pediatric-world.com/fukujin/p05.html) - Baquedano MS, et al. J Clin Endocrinol Metab 2013; 98: E153-161
- Ishii T, et al. J Endocrine Soc 2019; 3: 1367-1374
- Ishii T, et al. Eur J Endocrinol 2016; 175: 127-132
- Bose HS, et al. N Engl J Med 1996; 335: 1870-1878
- Ishii T, et al. J Clin Endocrinol Metab 2020; 105: 1870-1879
- Metherell LA, et al. J Clin Endocrinol Metab 2009; 94: 3865-3871
- Hatabu N, et al. J Clin Endocrinol Metab 2018; 104: 1866-1870
- Amano N, et al. Eur J Endocrinol 2017; 177: 187-194